
The brain initiates everything we do and processes all that we experience. Yet the mystery of the brain rivals that of the universe. In fact, we probably know more about the universe than we do about our own brains, which consist of approximately 100 billion neurons and 1 trillion support cells.
Elucidating how the brain works is a grand challenge within science. It will undoubtedly help us to better understand neurological diseases such as Alzheimer’s and Parkinson’s. Research in the last several decades has provided scientists with a good understanding of how neurons work, particularly at the levels of a single neuron or a small number of neurons.
With the help of magnetic resonance imaging (MRI), we also have some knowledge of brain activity over a large volume at the time scale of seconds to minutes. What we now need is technology to bridge the large gap in our understanding between microscopic interactions at the neuronal level and the macroscopic structures that perform complex computations.
|
Optical imaging is noninvasive and provides the high spatial resolution necessary to resolve individual neurons and neuronal processes. However, acquiring images through significant depths of the brain is no easy task since brain tissue is extremely heterogeneous, resulting in strong scattering by the various tissue components. Optical imaging technology will be essential in addressing these challenges, and it will feature prominently in U.S. President Obama’s recently announced BRAIN initiative (see sidebar).
Multiphoton microscopy (MPM), which was demonstrated more than two decades ago, has dramatically extended the depth penetration of high-resolution optical imaging. It has been a game-changer for neuroscience. When combined with genetically engineered fluorescent probes, MPM enables the non-invasive, dynamic measurement of neurons in their native environment. (See, for example, the review paper by J.N.D. Kerr and W. Denk.) It is well positioned to play a major role in expanding our understanding of brain function.
MPM for deep tissue imaging
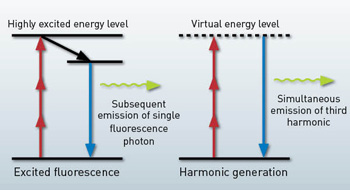
In MPM, image contrast is generated from nonlinearly excited fluorescence or harmonic generation. For fluorescence, a fluorophore is excited to an upper-energy level by the simultaneous absorption of multiple photons, and it subsequently emits a single fluorescence photon. The fluorescence lifetime is typically on the order of nanoseconds.
For harmonic generation, on the other hand, only virtual energy levels are involved, and emission is practically instantaneous. Femtosecond lasers are usually required for MPM to enhance the nonlinear excitation probability and hence the signal level. Compared to one-photon confocal microscopy, the deep tissue imaging capability of MPM mainly derives from nonlinear and longer-wavelength excitation.
First, nonlinear excitation confines the signal to the focal region (i.e., the excitation confinement), which enables 3-D sectioning without using a confocal pinhole in the signal path. Thus, with MPM, one can still efficiently collect signals generated deep within scattering tissue. Second, a longer wavelength is used in MPM to excite fluorophores because the energies of two or three photons are combined to generate the molecular transition. The longer excitation wavelength significantly reduces the effect of tissue scattering. The combined effect of a longer excitation wavelength and efficient signal collection makes MPM the method of choice for imaging the activity of individual neurons deep inside an intact brain.
Researchers have used two-photon microscopy (2PM) extensively for deep imaging in animal studies that delve into the mouse brain. Recently, a record imaging depth of 1.6 mm was attained in the cortical tissue, or gray matter, of a mouse brain in vivo (D. Kobat et al., 2011). The question then arises: What prevents deeper imaging?
Tissue absorption and scattering lead to the exponential decay of excitation light at the focus with increased imaging depth. Absorption is easy to determine, as it follows Beer’s law, which relates it to the properties of the material through which the light travels.
However, scattering is the dominant factor in reducing the excitation intensity at the focus. It causes photons to deviate from their ballistic path; deep within tissue, these scattered photons have essentially zero probability of reaching the focal volume. Thus, only the so-called ballistic photons can reach the geometrical focus.
 Three-photon microscopy. In three-photon microscopy, three photons are converted to a fluorescence photon or third harmonic photon. Compared with single photon excited fluorescence, multiphoton excited fluorescence is confined to the focus only due to the nonlinear excitation and enables 3-D sectioning in imaging.
Three-photon microscopy. In three-photon microscopy, three photons are converted to a fluorescence photon or third harmonic photon. Compared with single photon excited fluorescence, multiphoton excited fluorescence is confined to the focus only due to the nonlinear excitation and enables 3-D sectioning in imaging.
If the power deposited on the sample surface is below the tissue damage threshold, the signal decay from absorption and scattering can be compensated by raising the input power exponentially as depth increases. However, there is a fundamental limit that determines the maximum imaging depth for 2PM, which was shown by Theer et al. in 2003. The exponential decay of the excitation intensity at the focus when imaging deep into a scattering tissue causes nonlinear excitation to be no longer confined to the focal volume.
A quantitative measure of the 3-D excitation confinement is the signal-to-background ratio (SBR), which is simply the ratio of the amount of excitation within the focal volume to the amount outside. When this SBR approaches unity, the signal generated at the focus is overwhelmed by the background; hence, no useful feature can be resolved at the focus. Indeed, it is the SBR, rather than the decreasing signal strength, that ultimately limits the maximum penetration depth in 2PM. For example, this depth limit for 2PM in the mouse neocortex at 775-nm and 1,280-nm excitation is about 700 and 1,600 µm, respectively.
3PM for deeper imaging
The depth limit set by the SBR strongly suggests that deeper brain imaging requires tighter excitation confinement. Three-photon microscopy (3PM) was demonstrated in the mid-1990s, mainly to extend the spectral range of existing lasers. For example, researchers used femtosecond pulses generated by titanium:sapphire (Ti:S) oscillators to excite intrinsic molecules such as serotonin. However, femtosecond lasers with modest pulse energy (e.g., the Ti:S oscillator) are not well-suited for three-photon excitation (3PE), and thus three-photon fluorescence microscopy was generally considered a curiosity rather than a practical tool.
 Two-photon microscopy depth limit. The signal-to-background ratio limits the maximum penetration depth in 2PM. In the mouse neocortex, the depth limit at 775-nm and 1,280-nm excitation is about 700 and 1,600 µm, respectively.
Two-photon microscopy depth limit. The signal-to-background ratio limits the maximum penetration depth in 2PM. In the mouse neocortex, the depth limit at 775-nm and 1,280-nm excitation is about 700 and 1,600 µm, respectively.
The pursuit of a tighter excitation confinement for deep-tissue imaging provides a clear motivation for 3PM. Simple math shows that, away from the focus, signal decays approximately as 1/z2 and 1/z4 for 2PM and 3PM, respectively. 3PM has a much better signal confinement than 2PM because it suppresses the out-of-focus background. This shows that, for the same imaging depth, 3PM has a higher SBR and can be used to image deeper than 2PM. Indeed, within an imaging depth of 2.5 mm in brain tissue, SBR does not appear to limit 3PM.
Excitation wavelength for deep-tissue imaging
As we stated earlier, attenuation of excitation leads to SBR degradation in deep-tissue imaging. The smaller the attenuation, the greater the imaging depth that can be obtained before SBR reaches unity. What wavelength attenuates least in brain tissue?
A previous 2PM experiment of in vivo brain tissue shows that a 1,280-nm excitation wavelength can penetrate deeper than a 775-nm excitation wavelength (D. Kobat et al., 2011). Excitation attenuation is characterized by the effective attenuation length (le=(1/ la +1/ ls)–1), which factors both the tissue absorption length (la) and scattering length (ls). At 1,280 nm (285 μm), le is approximately twice what it is at 775 nm (131 μm) due to the reduced scattering at the longer wavelength.
At first glance, it seems that longer excitation wavelengths will always correlate to greater imaging depth due to reduced tissue scattering. Unfortunately, this is not true, since water absorption increases dramatically in the infrared, and roughly 77 percent of the brain is comprised of water. Thus, the optimum wavelength for deep imaging is the result of a tradeoff between tissue absorption and scattering. The optimum excitation wavelength (i.e., the wavelength for the largest le) appears to be near 1,700 nm, leveraging the relatively transparent window of water absorption between 1,600 and 1,800 nm. With 3PM at this spectral window, many orange or red fluorophores can be excited, including red fluorescent proteins (RFPs).
Laser construction for 1,700-nm 3PM
As with any MPM, the first step in performing 1,700-nm 3PM is to select a proper femtosecond laser. So far, the most popular laser source is the femtosecond mode-locked Ti:S oscillator, with a tuning range from 690 nm to 1,100 nm. You can further extend the wavelength tunability from 1,100 to 2,000 nm by synchronously pumping optical parametric oscillators (OPO) using a Ti:S laser. The combination of Ti:S and OPO can cover the excitation spectrum relevant to MPM. However, at the 1,700-nm window for deep-tissue imaging, the pulse energy delivered by OPOs is typically well below 10 nJ, which is not suitable for deep-tissue 3PM.
In order to build a compact, robust and economical laser source at 1,700 nm while delivering the high-pulse-energy and ultrashort pulse width needed for deep-tissue 3PM, we resort to soliton self-frequency shift (SSFS) in a special waveguide called a photonic crystal (PC) rod; it has an exceptionally large mode area (LMA) and is pumped by a commercial high-pulse-energy 1,550-nm fiber laser—a compact, turnkey system of 130 × 48 × 18 cm (L × W × H) in total.
|
|---|
 Energy scaling: An increase in the effective mode area (Aeff) leads to proportional increase in soliton energy.
Energy scaling: An increase in the effective mode area (Aeff) leads to proportional increase in soliton energy.
Optical solitons in optical fibers and SSFS
You can generate an optical soliton by balancing the linear anomalous group velocity dispersion (GVD, which induces pulse broadening when acting alone) and the nonlinear self-phase modulation, which induces spectral broadening when acting alone. Conventional silica fibers have anomalous dispersion above 1,300 nm, while photonic crystal fibers (PCFs), such as index-guided PCFs or air-core photonic bandgap fibers (PBGFs), can be anomalous below 1,300 nm and even in the visible wavelength. Optical solitons have superb pulse quality and the temporal intensity profile is a sech2 function.
Demonstrated in 1986 by F.M. Mitschke et al., SSFS occurs due to the intra-pulse stimulated Raman scattering effect, which continuously transfers the pulse energy from the higher frequency (shorter wavelength) to the lower frequency (longer wavelength). The red-shifting of solitons by hundreds of nanometers is possible through SSFS in an optical fiber.
Energy scaling for optical solitons
To obtain a high-energy soliton for deep-tissue 3PM, we need to know how to scale the soliton energy. The energy of a fundamental soliton is given by
 |
|---|
where c is light speed in vacuum, Aeff is the effective mode area, β2 is GVD, ω0 is the carrier frequency, n2 is nonlinear refractive index of the fiber core, and T0 is a measure of the pulse width. The energy formula implies that soliton energy is proportional to Aeff ; therefore, an effective way of increasing the soliton energy is by simply raising the Aeff .
The pulse energies that support stable Raman-shifted solitons in standard single-mode fibers (SSMFs) are approximately 1 nJ, which is too low for many practical applications. On the other hand, in air-core PBGFs, wavelength tunability is limited by the bandwidth of the bandgap to only tens of nanometers—although soliton energy on the order of 100 nJ can be obtained due to the much smaller n2 of air (a thousand times smaller than fused silica). LMA fibers and PC rods typically have an Aeff ranging from 256 to 4,600 μm2, which is3 to 60 times higher than SSMF.
So, the corresponding soliton energy is approximately 3 to 60 nJ. Furthermore, one can achieve wavelength tunability of hundreds of nanometers when the LMA fiber or the PC rod is pumped at 1,550 nm. As a result, with a commercial 1,550-nm high-energy femtosecond laser, one can use SSFS to generate a high-energy soliton at the 1,700-nm window. In a 36-cm PC rod with Aeff =2,300 μm2, we obtained 67-nJ, 65-fs soliton output with a center wavelength at 1,675 nm, suitable for deep-tissue 3PM (N.G. Horton et al., 2013).
Deep imaging of neurons in vivo with 1,700-nm 3PM
Using this high-energy, ultrafast laser system at the 1,700-nm spectral window, we performed 3PM of a mouse brain in vivo. 3PM can be performed with any multiphoton laser-scanning microscope. Special care must be taken to optimize the performance of the microscope system at 1,700 nm in order to minimize transmission loss. We used two photomultiplier tubes with proper filters to detect the 3PE fluorescence and third-harmonic generation (THG).
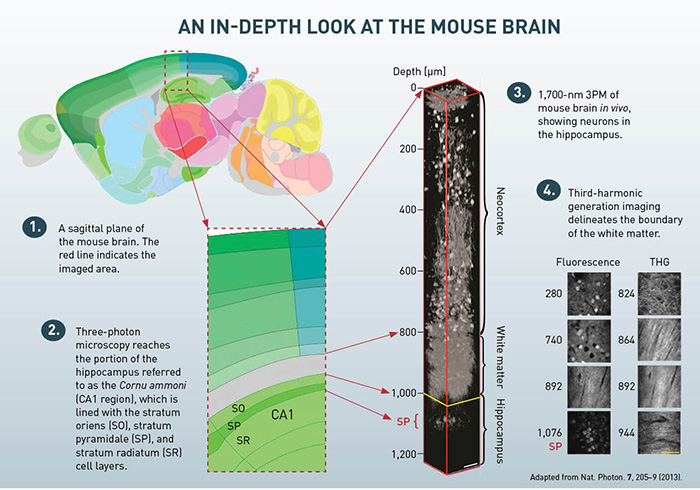
In our experiments, we performed a craniotomy and sealed it with cover glass to expose the brain, a routine procedure for in vivo MPM. The optical power on the sample was increased with imaging depth. In a first experiment, we used 3PM to image the hippocampus. This region of the brain is essentially impossible to image using 2PM due to the layer of white matter that separates it from the neocortex. THG images from 840 to 956 μm below the brain surface clearly reveal the white matter region and indicate that we achieved an imaging depth of about 400 μm into the hippocampus (roughly 1,360 μm below the surface of the brain).
We also performed 3PM in genetically modified mice whose neurons were labeled with RFP. Imaging clearly reveals the neurons in the stratum pyramidale (SP), a dense layer of pyramidal neurons within the hippocampus. We clearly resolved individual pyramidal neurons. Previous methods to reach the SP involved either removing overlying brain tissue (D.A. Dombeck et al., 2010) or inserting optical probes (M.J. Levene et al., 2004, and J. Jung et al., 2004).
To quantify the attenuation length, we used le, a standard method for measuring the fluorescence signal decay as a function of depth. The signal decay can be fit by an exponential function down to a depth of 840 μm (i.e., within the neocortex), yielding a measured le of 400 μm in the neocortex. The measured attenuation length in the white matter is 229 μm, which is much shorter than it is in the neocortex, presumably due to the highly scattering nature of the densely packed, myelinated axons.
 |
|---|
Perspective
MPM has changed how we visualize neurons by providing high-resolution, noninvasive imaging deep within brain tissue. Further improving the penetration depth is a major thrust of future development. Deep-tissue MPM is inherently multidisciplinary, so individuals with expertise in a variety of fields will contribute to this endeavor. The development of new laser systems, fiber optics, imaging optics and optoelectronics will continue to play a critical role.
While 3PM has significantly improved the depth penetration of MPM, it will be challenging to image significantly beyond 2-mm at a speed that is compatible with measuring neuronal function. Thus, new imaging methods, perhaps along the lines of dynamic compensation of tissue scattering, must be developed.
A major goal in brain imaging—and the BRAIN initiative—is to noninvasively record dynamic activity at the cellular level across a large area and depth in living animals. Fulfilling this goal, along with the development of a suite of tools for manipulating neural activity, will dramatically advance our understanding of how the brain works at the level of complex neural networks.
Although high-res optical imaging of brain activity will not be applicable for humans in the foreseeable future, the diversity and availability of animal models—which are largely modifiable through genetic engineering—are perhaps better suited for deriving basic knowledge about how neural circuits work. By enabling us to record brain activity from a large number of neurons and interacting neural circuits, optical imaging may elucidate the connections between neuron activities and behavior, illuminating one of the most profound mysteries in science.
Ke Wang is with the Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, China. Nicholas G. Horton and Chris Xu are with the School of Applied and Engineering Physics at Cornell University in Ithaca, N.Y., U.S.A.
References and Resources
-
F.M. Mitschke et al. Opt. Lett. 11, 659 (1986).
-
W. Denk et al. Science 248, 73 (1990).
-
J. Jung et al. J. Neurophysiol. 92, 3121 (2004).
-
M.J. Levene et al. J. Neurophysiol. 91, 1908 (2004).
-
J.N.D. Kerr et al. Nat. Rev. Neurosci. 9, 195 (2008).
-
D. Kobat. Opt. Express 17, 13354 (2009).
-
D.A. Dombeck et al. Nat. Neurosci. 13, 1433 (2010).
-
D. Kobat et al. J. Biomed Opt. 16, 106014 (2011).
-
N.G. Horton et al. Nat. Photon 7, 205 (2013).