
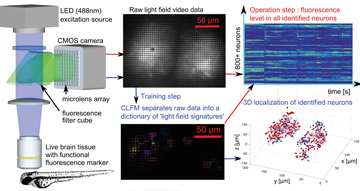
 In compressive light-field microscopy (CLFM), a light-field microscope first records neural activity in live brain tissue using genetically encoded neurons that express a functional fluorescence marker. Our method enables single-shot localization of active neurons in 3-D through scattering tissue. A training step processes raw data to build a dictionary of each neuron’s light-field signature. This dictionary enables decomposition of later fluorescence measurements into the set of active neurons in a single frame. Neural activity can then be quantified in the entire volume at video-frame rates without ever reconstructing a volume image.
In compressive light-field microscopy (CLFM), a light-field microscope first records neural activity in live brain tissue using genetically encoded neurons that express a functional fluorescence marker. Our method enables single-shot localization of active neurons in 3-D through scattering tissue. A training step processes raw data to build a dictionary of each neuron’s light-field signature. This dictionary enables decomposition of later fluorescence measurements into the set of active neurons in a single frame. Neural activity can then be quantified in the entire volume at video-frame rates without ever reconstructing a volume image.
Mapping and deciphering the living brain’s activity requires 3-D imaging with high temporal resolution, for simultaneously tracking thousands of individual neurons in a volume. Light-field microscopy (LFM) based on optogenetic activity is a promising solution that enables in vivo single-shot 3-D acquisition by capturing both the light’s intensity and its propagation direction.1 Yet traditional LFM relies on the intermediate reconstruction of visual renderings, followed by digital analysis.2 This limits neuroscience applications in deeper layers of the brain, as optical scattering destroys image quality.3
We have developed an LFM technique that eliminates the need to reconstruct a volume image.4 Instead, we detect and localize neurons by leveraging the sparsity of brain activity in space and time for better performance in highly scattering environments.5 We combine LFM hardware with compressed-sensing algorithms and wave-optical light-field scattering models to separate neurons. An initial training step identifies a unique light-field signature for each active neuron in the field of view, representing what the sensor would detect if only this neuron were activated. The signature includes the effects of optical scattering and allows accurate 3-D localization of the corresponding neuron.
Once all neurons within the field of view have been detected, we can decompose any new frame acquired into the basis set of optical signatures. The coefficients of this decomposition represent a time-stamped quantitative measurement of fluorescence in all neurons within the monitored volume.
Our technology tackles the optical-scattering issue and enables fast recording of neural activity through deep brain tissue. This process provides neuroscientists with a directly usable, quantitative result without ever reconstructing a volume image (see video).
By avoiding the intermediate step, our method also saves time and computer memory. The system and computational hardware can be miniaturized into compact implants for real-time implementations and online readout of neural activity. We believe that the approach we’ve developed significantly advances recording technology, and could allow for key breakthroughs in understanding and interacting with the brain.
Researchers
Nicolas C. Pégard, Hsiou-Yuan Liu, Nicholas Antipa, Maximillian Gerlock, Hillel Adesnik and Laura Waller, University of California, Berkeley, Calif., USA
References
1. M. Levoy et al. ACM Trans. Graph. 25, 924 (2006).
2. M. Broxton et al. Opt. Express 21, 25418 (2013).
3. R. Prevedel et al. Nat. Methods 11, 727 (2014).
4. N.C. Pégard et al. Optica 3, 517 (2016).
5. H.Y. Liu et al. Opt. Express 23, 14461 (2015).